
вЛЁЂЛљБОаХЯЂ
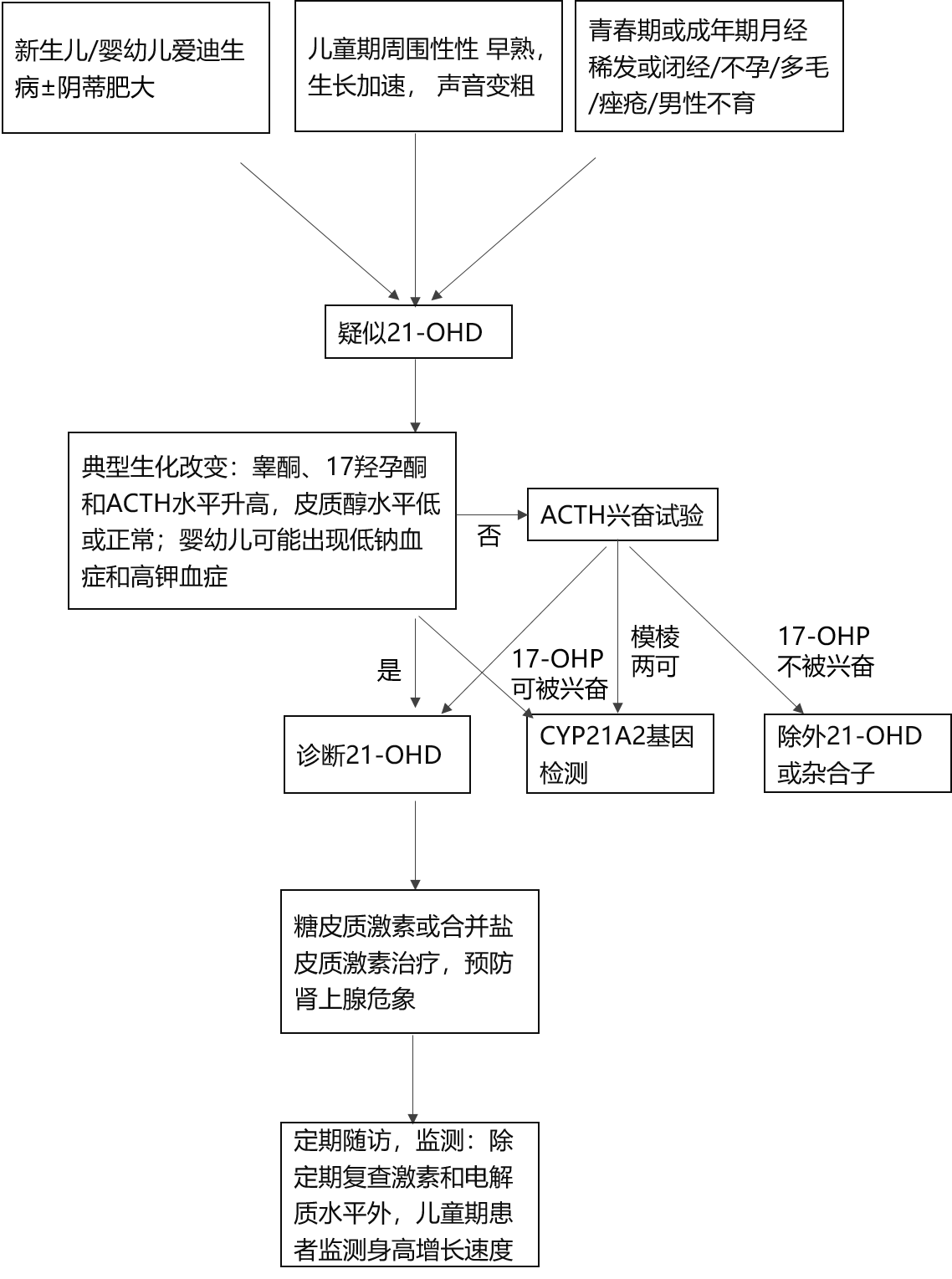
ИХЪіЃК21-єЧЛЏУИШБЗІжЂЃЈ21-hydroxylase deficiencyЃЌ21-OHDЃЉЪЧЯШЬьадЩіЩЯЯйдіЩњжЂЃЈcongenital adrenal hyperplasiaЃЌCAHЃЉжазюГЃМћЕФРраЭЃЌЪЧгЩгкБрТы21-єЧЛЏУИЕФCYP21A2ЛљвђШБЯнЕМжТЩіЩЯЯйЦЄжЪРрЙЬДММЄЫиКЯГЩеЯАЕФвЛжжЯШЬьадМВВЁЃЌГЪГЃШОЩЋЬхвўадвХДЋЁЃОЕфаЭЛМепПЩЗЂЩњЩіЩЯЯйЮЃЯѓЃЌЕМжТЩњУќЮЃЯеЃЛИпалМЄЫибЊжЂЪЙХЎадФаадЛЏЃЌЕМжТЙЧСфМгЫйНјеЙЁЂАЋЩэВФвдМАЧрДКЗЂг§вьГЃЃЌВЂгАЯьЩњг§ФмСІЁЃ
ВЁвђЃК21-OHDгЩЮЛгкШОЩЋЬх6p21.3ЧјгђФкЕФCYP21A2ЛљвђЭЛБфв§Ц№ЁЃЦфБрТыЕФЕААзЮЊ21єЧЛЏУИЃЈP450c21ЃЉЁЃИУУИДпЛЏ17єЧдаЭЊЃЈ17-OHPЃЉзЊЛЏЮЊ11-ЭббѕЦЄжЪДМЃЌЭЌЪБДпЛЏдаЭЊзЊЛЏЮЊ11-ЭббѕЦЄжЪЭЊЃЌЖўепЗжБ№ЮЊЦЄжЪДМКЭШЉЙЬЭЊЕФЧАЬхЁЃ21єЧЛЏУИЛюадНЕЕЭжТЦЄжЪДМКЭШЉЙЬЭЊКЯГЩЪмЫ№ЁЃЦЄжЪДМЫЎЦНКЯГЩМѕЩйЃЌЭЈЙ§ИКЗДРЁЪЙДЙЬхACTHЗжУкдіМгЃЌДЬМЄЩіЩЯЯйЦЄжЪЯИАћдіЩњЃЛЖјШЉЙЬЭЊЗжУкВЛзуМЄЛюЩЯгЮЩіЫиКЭбЊЙмНєеХЫиЂђЕФЗжУкЃЌЭЌЪБгЩгкжаМфВњЮяЕФЖбЛ§ЃЌЮЊадМЄЫиЃЈдкЩіЩЯЯйЦЄжЪжївЊЮЊалМЄЫиЃЉКЯГЩЬсЙЉСЫвьГЃдіЖрЕФЕзЮяЃЌВњЩњСЫХдТЗДњаЛПКНјЕФЬиеїадКѓЙћЁЊЁЊИпалМЄЫибЊжЂЁЃалМЄЫиЩ§ИпЯджјГЬЖШвРДЮЮЊалЯЉЖўЭЊЁЂиКЭЊКЭЭбЧтБэалЭЊЃЈDHEAЃЉЁЃ
CYP21A2ЛљвђЕФЭЛБфРраЭгаАйгржжЃЌ80ЃЅДцдкЛљвђаЭКЭБэаЭЕФЯрЙиадЁЃЕБЭЛБфЕМжТ21єЧЛЏУИЛюадЕЭгк1ЃЅЪБЃЌБэЯжЮЊбЯжиЪЇбЮЃЌГЪЯжЕЭФЦбЊжЂКЭИпМибЊжЂЃЌаТЩњЖљЩіЩЯЯйЮЃЯѓЁЃЕБУИЛюадВаСєЮЊ1ЃЅЁЋ2ЃЅЪБЃЌШЉЙЬЭЊЛЙПЩЮЌГждке§ГЃЗЖЮЇЃЌЪЇбЮЧуЯђЕЭЃЈЕЋгІМЄЪБШдПЩЗЂЩњЃЉЁЃУИЛюадБЃСєга20ЃЅЁЋ50ЃЅЪБЃЌЦЄжЪДМКЯГЩМИКѕВЛЪмЫ№ЁЃАДЛљвђаЭ-СйДВБэаЭЕФЙиЯЕЁЂШЉЙЬЭЊКЭЦЄжЪДМШБЗІЕФГЬЖШЁЂИпалМЄЫиЕФбЯжиГЬЖШЃЌ21-OHDЗжЮЊСНДѓРраЭЃК
1.ОЕфаЭ21-OHDЃЌАДШЉЙЬЭЊШБЗІГЬЖШгжЗжЮЊЪЇбЮаЭЃЈsalt wastingЃЌSWЃЌдМеМ75ЃЅЃЉКЭЕЅДПФаадЛЏаЭЃЈsimple virilizingЃЌ SVЃЌдМеМ25ЃЅЃЉЃЛ
2.ЗЧОЕфаЭ21-OHDЃЈnon classical CAHЃЌNCCAHЃЉЁЃ
СїааВЁбЇЃКCAHИљОнШБЯнУИЕФжжРрПЩЗжЮЊЖржжРраЭЃЌ21-OHDЪЧзюГЃМћЕФРраЭЃЌеМ90ЃЅЁЋ95ЃЅЁЃЙњФкЭтЕФЖрЪ§баОПЬсЪОаТЩњЖљЩИВщЗЂВЁТЪЮЊ1/20000ЁЋ1/10000ЁЃ
ЖўЁЂМВВЁеяЖЯ
СйДВБэЯжЃК21-OHDЕФСйДВБэЯжАќРЈВЛЭЌГЬЖШЕФЪЇбЮКЭИпалМЄЫибЊжЂСНДѓРрЁЃаТЩњЖљЦ№ВЁЕФЛМЖљБэЯжЮЊВЛЭЌГЬЖШЩіЩЯЯйЦЄжЪЙІФмВЛзуЕФБэЯжЃЌШчШэШѕЮоСІЁЂЖёаФЁЂХЛЭТЁЂЮЙбјРЇФбЁЂИЙаКЁЂТ§адЭбЫЎЁЂЦЄЗєЩЋЫиГСзХКЭЩњГЄГйЛКЕШЁЃЩіЩЯЯйЮЃЯѓГЃЪЧ21-OHD SWаЭдкаТЩњЖљЦкЕФЪзЗЂБэЯжЃЌБэЯжЮЊбЯжиЕЭбЊФЦЁЂИпбЊМиЁЂЕЭбЊШнСПаданПЫЃЌПЩАщгаЕЭбЊЬЧЃЌгЩгІМЄгеЗЂЁЃбЯжиЕФЕЭФЦбЊжЂПЩЕМжТГщДЄЕШжаЪрЩёОЯЕЭГБэЯжЃЌбЯжиЕФИпМибЊжЂдђПЩв§Ц№жТУќЕФаФТЩЪЇГЃЁЃ
ИпалМЄЫибЊжЂдкВЛЭЌФъСфБэЯжВЛвЛЁЃОЕфаЭ21-OHDЕФХЎадЛМЖљГіЩњЪБЭтЩњжГЦїгаВЛЭЌГЬЖШФаадЛЏЁЃЧсепБэЯжЮЊЙТСЂадвѕЕйЗЪДѓЃЌжиепЭтЩњжГЦїПЩНгНќФаадЃЌДѓвѕДНГЪвѕФвбљЃЌвѕЕйГЪФђЕРЯТСбаЭвѕОЅбљЃЌВЂОпЙВЭЈЕФФђЕРвѕЕРПкЁЃЕЋДѓвѕДНФкВЛФмДЅМАадЯйЃЌгаЭъШЋе§ГЃЕФХЎадФкЩњжГЦїНсЙЙЃЈТбГВКЭзгЙЌЃЉЁЃФаадаТЩњЖљЦкКЭгЄЖљЦкЪБПЩФмЮовѕОЅдіДѓЕШЭтЩњжГЦївьГЃЃЌЪЧбгЮѓеяЖЯЕФГЃМћдвђЁЃжСгзЖљЦкЃЌСНадОљЛсГЪЯжЭтжмададдчЪьЁЃФаКЂЛМЖљГЪЯжвѕОЅдіДѓЃЌАщЛђВЛАщвѕУЋдчЯжЃЛХЎадЛМЖљГЪЯжвьададдчЪьЁЃГЄЦкИпЫЎЦНадМЄЫиДЬМЄЯТЧ№ФдДйадЯйМЄЫиЪЭЗХМЄЫиЃЈGnRHЃЉЩёОдЊЃЌЗЂеЙЮЊжаЪрададдчЪьЁЃХЎадЛЙПЩгаЕкЖўадеїЗЂг§ВЛСМКЭдЗЂадБеОЛђдТОЯЁЗЂЁЃ21-OHDЗЧОЕфаЭдкЧрДКЦкЛђГЩФъКѓБЛФтеяЮЊЖрФвТбГВзлКЯеїЪБВХБЛШЗеяЮЊ21-OHDЁЃСНадОљдкгзФъЦкПЊЪМЗЂЩњЯпадЩњГЄАщЙЧСфдіГЄМгЫйЃЌЪЙжеЩэИпЪмЫ№ЁЃ
ЦфЫћБэЯжАќРЈЦЄЗєКЭ№ЄФЄЩЋЫидіЩюЃЌвдШщдЮКЭЭтвѕЮЊЯдЃЌВПЗжЛМЖљПЩЮоДЫИФБфЁЃ
NCCAHЖљЭЏЦкКЭЧрДКЦкЩѕжСГЩФъЦкСйДВГЪВЛЭЌГЬЖШЕФИпалМЄЫибЊжЂБэЯжЃЌвВгаНіБэЯжЮЊЩњГЄМгЫйКЭЙЧСфПьЫйНјеЙЁЃ
ИЈжњМьВщЃК
1.бЊЧх17-OHP
17-OHPЩ§ИпЪЧ21-OHDЕФЬивьадеяЖЯжИБъКЭжївЊжЮСЦМрВтжИБъЁЃвЛАуЖјбдЃЌ17-OHPЩ§ИпЗљЖШдНИпЃЌУИШБЯнГЬЖШдНжиЁЃЕЋ17-OHPгыACTHвЛбљЖМЪЧгІМЄМЄЫиЃЌвђДЫдкгагІМЄЕФЧщПіЯТЃЌЛђЛМЖљГщбЊПоФжбЯжиЪБЖМПЩНЯЪЕМЪЫЎЦНгаУїЯдЕФЩ§ИпЃЌНтЖСНсЙћЪБашПМТЧЕНДЫжжвђЫиЁЃ
2.ЛљДЁбЊЧхЦЄжЪДМКЭACTH
ОЕфаЭЛМепбЊЧхЦЄжЪДМНЕЕЭАщACTHЩ§ИпЁЃвВга21-OHDЛМепЦЄжЪДМдке§ГЃЗЖЮЇЃЌЖјACTHЩ§ИпЃЌашНсКЯЦфЫћжИБъзлКЯХаЖЯЁЃ21-OHDЗЧОЕфаЭЛМепСНжжМЄЫиЛљБОдке§ГЃЗЖЮЇЁЃ
3.алМЄЫи
ИїалМЄЫиВтЖЈжЕашАДееадБ№ЁЂФъСфКЭЧрДКЗЂг§ЦкНЈСЂЕФе§ГЃВЮеежЕХаЖЯЁЃалЯЉЖўЭЊгы17-OHPгаНЯКУЕФЯрЙиадЃЌеяЖЯКЭМрВтвтвхзюМбЁЃDHEASдкЖрФвТбГВзлКЯеїжаврПЩгаЩ§ИпЁЃ
4.бЊНЌЩіЫиХЈЖШЛђЩіЫиЛюадЁЂбЊЙмНєеХЫиЂђКЭШЉЙЬЭЊ
ЩіЫидк21-OHD SWаЭЩ§ИпЃЌЕЋеяЖЯЬивьадВЛИпЁЃВПЗжЗЧЪЇбЮаЭЛМепЩіЫивВПЩЩ§ИпЃЌЩіЫиЪЧбЮЦЄжЪМЄЫиЬцДњжЮСЦжаЕФживЊМрВтжИБъЁЃШЉЙЬЭЊЕЭЯТжЇГжЪЇбЮаЭеяЖЯЃЌЕЋга1/4ЛМЖљбЊЧхШЉЙЬЭЊПЩе§ГЃЁЃ
5.ШОЩЋЬх
ШОЩЋЬхжївЊгУгкГ§Эт46ЃЌXYадЗЂг§вьГЃМВВЁЁЃ
6.гАЯёбЇ
ЩіЩЯЯйЕФBГЌКЭCTЕШгАЯёбЇМьВщгажњгкЩіЩЯЯйжзСіЛђЦфЫћЩіЩЯЯйЃЈЗЂг§ВЛСМЃЉВЁБфМјБ№ЁЃХЎадгІЭъЩЦзгЙЌЁЂЫЋИНМўBГЌЃЌ2ЫъПЊЪМашМьВщЙЧСфЁЃ
7.ЛљвђеяЖЯ
21-OHDЕФЛљвђеяЖЯЮоТлЖдЩњЛЏеяЖЯУїШЗЛђепВЛУїШЗЕФМјБ№еяЖЯЖМЪЎЗжживЊЃЌВЂЧвФмеяЖЯдгКЯзгаЏДјепЃЌЖдвХДЋзЩбЏвВЗЧГЃживЊЁЃ
еяЖЯЃК21-OHDеяЖЯашзлКЯСйДВБэЯжЁЂАќРЈ17-OHPдкФкЕФИїЯрЙиМЄЫиХЈЖШРДМгвдХаЖЯЃЌЛљвђМьВтПЩНјвЛВНУїШЗеяЖЯЁЃФПЧАЙњФкНЯЖрЕиЧјвбПЊеЙ21-OHDЕФаТЩњЖљЩИВщЃЌЖдгкзуИњбЊЩИВщ17-OHPбєадепЃЌашАДееЩИВщЙВЪЖВйзїЁЃ
NCCAHЛМепбЊЧхЦЄжЪДМе§ГЃЛђдке§ГЃЯТЯоЃЌACTHе§ГЃЛђСйНчИпжЕЁЃгУ17-OHPЛљДЁжЕеяЖЯОпВЛШЗЖЈадЃЌЛљвђМьВтМЋЦфживЊЁЃ
МјБ№еяЖЯЃКГЃМћЕФашвЊгы21-OHDМјБ№еяЖЯЕФМВВЁАќРЈЃК
1.11ІТ-єЧЛЏУИШБЯнЃЈCYP11B1ЛљвђЭЛБфЃЉ
вВгаИпалМЄЫибЊжЂЃЌМЋЩйгаГіЩњЪБЪЇбЮБэЯжЃЌГЃМћбЮЦЄжЪМЄЫиЙ§ЖрШчЫЎФЦфѓСєЁЂЕЭбЊМиКЭИпбЊбЙЕШЃЌЩіЫи-бЊЙмНєеХЫиЫЎЦНЕЭЃЌдаЭЊгы17-OHPЩ§ИпЁЃЕЋВПЗжЛМепбЊбЙПЩе§ГЃЃЌБивЊЪБашааЛљвђМьВтгы21-OHDМјБ№ЁЃ
2.17ІС-єЧЛЏУИШБЯнжЂЃЈCYP17A1ЛљвђЭЛБфЃЉ
ДЫУИЭЌЪБЛЙОпга17ЃЌ20-СбСДУИЕФЛюадЃЌСйДВБэЯжЮЊбЮЦЄжЪМЄЫидіЖрЕФжЂзДЃЌШчЕЭбЊМиЁЂИпбЊбЙвдМАадМЄЫиВЛзуЕФБэЯжЃЌШчХЎадЧрДКЗЂг§ШБЪЇЃЌФаадХЎадЛЏЁЃдаЭЊЩ§ИпЃЌ17-OHPНЕЕЭЛђе§ГЃЁЃ
3.ЯШЬьадвХДЋадЩіЩЯЯйЗЂг§ВЛСМ
ЪЧгЩгкNR0B1ЛљвђЛђSF1ЛљвђЭЛБфЕМжТЕФЯШЬьадЩіЩЯЯйЦЄжЪЙІФмМѕЭЫЃЌПЩКЯВЂадЯйЙІФмЕЭЯТЃЌЦфгАЯёбЇЖрБэЯжЮЊЩіЩЯЯйЗЂг§ВЛСМЁЃ
4.ЩіЩЯЯйЦЄжЪжзСі
ЩіЩЯЯйЦЄжЪжзСіЃЈгШЦфЪЧЖљЭЏЃЉГЃвдИпалМЄЫибЊжЂЕФСйДВБэЯжЦ№ВЁЃЌАщЛђВЛАщЦЄжЪДМдіЖржЂЃЌЩѕжСга17-OHPЯджјЩ§ИпЃЌЕЋACTHУїЯдЕЭЯТЪЧМјБ№вЊЕуЁЃгАЯёбЇжЄЪЕеМЮЛВЁБфЁЃ
5.ЖрФвТбГВзлКЯеї
ЖдгкЧрДКЦкЛђГЩФъЦквђдТОЪЇЕїЛђИпалМЄЫибЊжЂОЭеяЕФХЎадЛМепЃЌNCCAHЕФБэЯжПЩгыЖрФвТбГВзлКЯеїгавЛЖЈжиЕўЃЌЧвЖрФвТбГВзлКЯеїврПЩГіЯжDHEASЕФЩ§ИпЃЌПЩЭЈЙ§жаМССПЕиШћУзЫЩвжжЦЪдбщМјБ№ЃЌБивЊЪБааCYP21A2ЛљвђМьВтвдУїШЗеяЖЯЁЃ
Ш§ЁЂжЮСЦЗНЪН
жЮСЦЃК
1.жЮСЦФПБъ
АДее21-OHDВЛЭЌРраЭжЦЖЈжЮСЦФПБъЁЃжЮСЦФПБъАќРЈЬцДњЩњРэашвЊСПЕФЬЧЦЄжЪМЄЫиЃЌЭЌЪБКЯРэвжжЦИпалМЄЫибЊжЂЃЌОЁПЩФмЛжИДе§ГЃЩњГЄЗЂг§ЕФЙьМЃЃЌДяЕНРэЯыЕФжеЩэИпЃЌИФЩЦдЖЦкЩњжГНЁПЕЁЃ
2.ЬЧЦЄжЪМЄЫижЮСЦ
ЧтЛЏПЩЕФЫЩЪЧЛљДЁгУвЉЃЌашвЊжеЩњЕФЬцДњжЮСЦЁЃНЈвщЗжБ№АДееЛМепЩадкЩњГЄжаКЭвбДяЕНГЩФъЩэИпЧщПіжЦЖЈЗНАИЁЃЮДЭЃжЙЩњГЄепЃЌНЈвщгУЧтЛЏПЩЕФЫЩЬцДњЁЃДяЕНГЩФъЩэИпКѓЃЌПЩвдИјАыЫЅЦкЯрЖдГЄЕФжЦМСЃЌШчЦУФсЫЩЛђЕиШћУзЫЩЁЃЧтЛЏПЩЕФЫЩЬцДњжЮСЦЗНАИашдкВЮеевЉЮяДњаЛЖЏСІбЇЕФддђЩЯНЈСЂИіЬхЛЏЗНАИЁЃИљОнФъСфЩшЖЈМССПЃЌЗжДЮИјвЉЃЌИљОнМрВтНјааМССПЕїНкЁЃЬцДњжЮСЦМССПКЭЗНАИгІНсКЯФъСфКЭЗЂг§ЦкИіЬхЛЏЩшЖЈЃЌВЂОЁПЩФмПижЦдкзюЕЭгааЇМССПЃЌБмУтЙ§СПЖдЩњГЄЕФвжжЦКЭЗЂЩњвНдДадПтаРзлКЯеїЁЃдкгІМЄзДЬЌКЭМВВЁЪБашЖдЬЧЦЄжЪМЄЫиЕФМССПНјааЕїећЁЃвЛАуЖљЭЏгУСПЧтЛЏПЩЕФЫЩ10ЁЋ15mg/m2ЃЌУПШеЗж3ДЮПкЗўЃЛГЩШЫгУСП15ЁЋ25mg/dЃЌУПШеЗж2ЁЋ3ДЮПкЗўЁЃ
3.бЮЦЄжЪМЄЫижЮСЦ
ашвЊдкЗРжЙЪЇбЮЮЃЯѓЭЌЪБЃЌЙизЂбЮМЄЫиУєИаадЕФФъСфБфЛЏЙцТЩМАЪБЕїећМССПЃЌБмУтЗЂЩњвНдДадИпбЊбЙЁЃ21-OHDЪЇбЮаЭдкЬЧЦЄжЪМЄЫиЛљДЁЩЯСЊгУбЮЦЄжЪМЄЫиПЩвдМѕЩйЬЧЦЄжЪМЄЫиЕФзмСПМАГЄЦкВЛСМЗДгІЁЃЗњЧтПЩЕФЫЩЪЧФПЧАЮЈвЛЕФбЮЦЄжЪМЄЫижЦМСЃЌПЩвдУПШеЗж1ЁЋ2ДЮЗўгУЃЌМССПАДЖдбЮЦЄжЪМЄЫиУєИаадЕФФъСфИФБфЙцТЩЩшжУЃЌИљОнМрВтНјааМССПЕїНкЁЃгЩгкжСНёЩаЮДгаХаЖЯСЦаЇЕФЕЅвЛМЄЫивдМАЙЬЖЈЕФЧаЕуЕФЁАН№БъзМЁБЃЌНЈвщашНсКЯМЄЫиКЭСйДВжИБъМрВтзлКЯХаЖЯЃЌЪЕЯжДяЕНИіЬхЛЏжЮСЦЕФзюРэЯыФПБъЁЃвЛАуЖљЭЏгУСПЗњЧтПЩЕФЫЩ0.05ЁЋ0.2mg/dЃЌУПШеЗж1ЁЋ2ДЮЗўгУЃЛГЩШЫМССП0.05ЁЋ0.2mg/dЃЌУПШе1ДЮПкЗўЁЃ
4.ЩњГЄМЄЫиКЭДйадЯйМЄЫиЪЭЗХМЄЫиРрЫЦЮя
ЖдгкаддчЪьбЯжиЃЌЙЧСфГЌЧАУїЯдЃЌдЄВтГЩФъЩэИпЫ№ЪЇНЯЖрепЃЌПЩПМТЧЩњГЄМЄЫижЮСЦЁЃЖдгквбОЗЂЩњжаЪрададдчЪьЕФЛМепЃЌПЩСЊКЯДйадЯйМЄЫиЪЭЗХМЄЫиРрЫЦЮяЁЃЕЋДЫРрвЉЮяЖдгкжеЩэИпЕФИФЩЦгыЛМепдЗЂВЁЕФПижЦЁЂЖдвЉЮяЕФжЮСЦЗДгІЁЂЖдЙЧСфГЌЧАЕФГЬЖШЁЂИИФИЕФвХДЋЩэИпЕШЖрЗНУцвђЫигаЙиЃЌвђДЫжЮСЦаЇЙћИіЬхВювьНЯДѓЁЃ
еяСЦСїГЬЭМЃК
ВЮПМЮФЯзЃК
[1] Speiser PW,Azziz R,Baskin LS,et al.Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab,2010,95(9):4133-4160.
[2] Fleming L,Van Riper M,Knafl K. Management of childhood congenital adrenal hyperplasia-an integrative review of the literature. J Pediatr Health Care,2017,31(5):560-577.
[3] Anzo M,Adachi M,Onigata K,et al. Guidelines for diagnosis and treatment of 21-hydroxylase deficiency (2014 revision).Clin Pediatr Endocrinol,2015,24(3):77-105.
[4] ДїКУ, ТЌСе, аЯаЁЦН, ЕШ. жаМССПЕиШћУзЫЩалМЄЫивжжЦЪдбщдкХЎадИпалМЄЫибЊжЂжаЕФеяЖЯМлжЕ. жаЛЊвНбЇдгжО,2018,98(26):2073-2077.
[5] жаЛЊвНбЇЛсЖљПЦбЇЗжЛсФкЗжУквХДЋДњаЛВЁбЇзщ. ЯШЬьадЩіЩЯЯйЦЄжЪдіЩњжЂ21-єЧЛЏУИШБЯнеяжЮЙВЪЖ. жаЛЊЖљПЦдгжО, 2016,54(8):569-576.
АцШЈЩљУї
вдЩЯФкШнРДздСМвНЛу-КБМћВЁаТНјеЙЃЌШчгаНЈвщЛђвЩЮЪЃЌЛЖгжТЕч18017449015ЁЃ
 ГЄАДЖўЮЌТы
ГЄАДЖўЮЌТыЙизЂОЋВЪФкШн





