患者女性,18岁时以脊髓炎和复视起病,随后经历多次复发。头颅MRI显示多发性硬化(MS)特征性T2病变(卵圆形白质病变和Dawson手指征),脑脊液显示免疫球蛋白指数升高和寡克隆区带阳性,被诊断为临床确定的复发缓解型多发性硬化,之后接受了多种多发性硬化疾病修正药物治疗,并逐渐进展为继发进展型多发性硬化(SPMS)。患者57岁时,出现急性长节段横贯性脊髓炎,且AQP4抗体阳性。经过对临床病程、影像学表现、分子诊断方法和治疗反应特征的分析最终认为,患者最有可能先后出现多发性硬化和视神经脊髓炎谱系疾病而并非误诊。该病例强调了神经学实践中的一个关键原则,即当出现新的发病特征时,应及时考虑到新增的不同诊断。
病例介绍
患者为一名57岁女性,在克利夫兰诊所多发性硬化(MS)诊疗中心接受数十年的随访,起初患者被临床确诊为复发缓解型多发性硬化(RRMS),随后进展为继发进展型多发性硬化(SPMS),目前患者又出现长节段横贯性脊髓炎的临床特征。1981年,患者18岁时出现横贯性脊髓炎表现及复视(图1),经过皮质类固醇治疗后症状完全缓解,被临床确诊为RRMS,随后接受干扰素β-1a的治疗,并在该种治疗下保持了近30年的临床神经病学稳定。
直到2009年,新的临床复发促使她的疾病修正治疗(DMT)药物过渡到醋酸格拉替雷。尽管在之后大约10年的时间里,服用醋酸格拉替雷使得患者病情得到相对稳定,没有出现疾病活动的临床证据,但在2019年5月的随访中显示,患者在信息处理速度、25英尺步行实验和疲劳等方面的测试中显示出疾病恶化。2019年5月的脑部MRI显示了MS的高度特征性病变(如Dawson手指征、卵圆形并垂直于脑室长轴的白质病变),以及深部白质中多个新的和/或扩大的T2高信号无强化病变(图2)。重新评估后发现,考虑该患者进展为SPMS。同年11月,患者出现亚急性发作的双侧肢体无力,导致无法行走,需要使用轮椅(扩展残疾状态量表评分为7分)。当时查体显示,上肢肌力正常,下肢肌力3级;振动觉减弱,左侧为著;腱反射增强但不伴有阵挛;指鼻试验欠稳准。口服大剂量泼尼松治疗后,再次出现明显改善,可借助助行器行走。
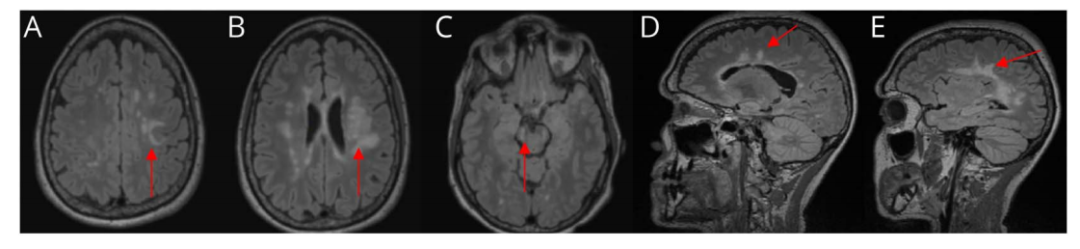
图2 患者2019年5月头颅MRI表现(箭头所指为病灶部位)
2020年2月,患者开始服用西尼莫得(一种口服鞘氨醇-1-磷酸(S-1-P)受体调节剂)。在服用西尼莫得一个月后,患者出现了行走困难、全身无力、头晕、构音障碍、痉挛及精神错乱。脑部MRI显示多个新的强化脑部病变,最显著的病灶位于右侧半卵圆中心、右侧侧脑室前方和上部的脑室周围(图3)。再次接受大剂量类固醇和抗生素治疗(尿路感染)。
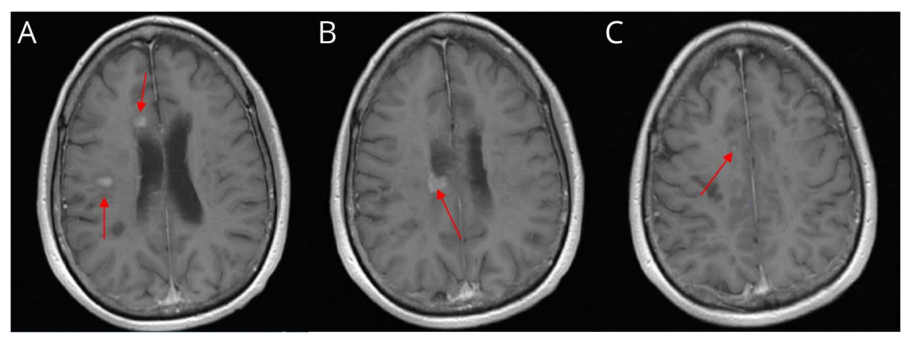
图3 患者2020年5月头颅MRI表现(箭头所指为病灶部位)
两个月后,新出现左上肢无力,并伴有双侧非持续性踝阵挛。因患者外周血淋巴细胞减少至200/μL因而维持原西尼莫得剂量。此时脑部MRI未显示新的异常强化病灶,也没有急性缺血的证据。颈椎和胸椎MRI显示不连续的短节段无强化病变。此次再次接受大剂量皮质类固醇和抗生素治疗(尿路感染),病情好转。然而,一周后,患者因步态恶化、吞咽困难和虚弱入院并进入重症监护病房接受气管插管和血管活性药物以维持血压。脑脊液分析显示免疫球蛋白指数升高及寡克隆区带阳性。后患者病情逐渐得到控制并进行积极康复,此时患者上肢仅存微量肌力并出现截瘫和尿潴留。考虑到病情恶化的严重性,患者接受了静脉注射甲强龙和一个疗程的血浆置换。复查脊髓成像显示从颈髓交界处到上胸脊髓的纵向广泛的弥漫性高信号病变,伴有周边强化和明显的水肿(图4)。经脑脊液抗体检测发现AQP4抗体滴度为1:2560,确诊为视神经脊髓炎谱系疾病(NMOSD)。此时停用西尼莫得,同时开始使用CD20单抗――奥瑞珠单抗。
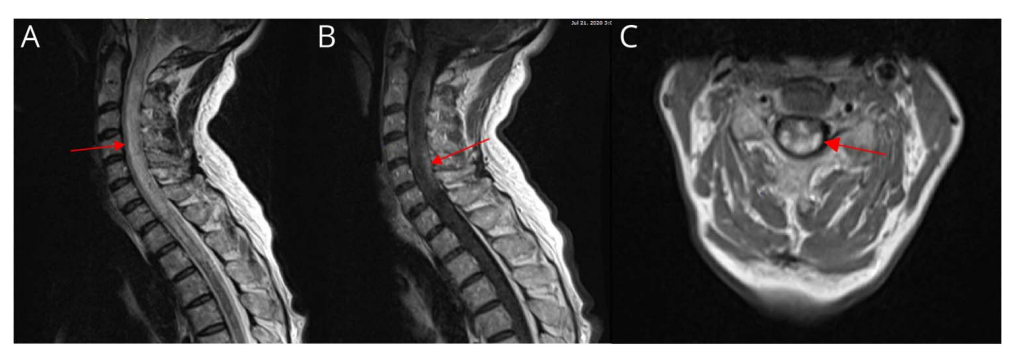
图4 患者2020年7月脊髓MRI表现(箭头所指为病灶部位)
病例讨论
这个病例有2种可能性:第一,患者最初被误诊为RRMS,而实际上是非典型发病的NMOSD,直到长截断脊髓炎的出现得以确诊。另一种可能是,她在时间上先后出现了两种不同的神经炎性疾病,即18岁时出现多发性硬化,57岁时出现NMOSD。
首先来看第一种情况:少数NMOSD病例在临床上也可以表现出与MS类似的短节段脊髓病变。此外,NMOSD患者可能出现有与MS一致的颅内病变。研究表明,60%的NMOSD患者可出现颅内白质病变,并且高达16%的NMOSD患者颅内病灶符合MS诊断中头颅MRI的Barkhof 标准。该病例对升级的DMTs药物缺乏反应,而患者的临床恶化可能由于西尼莫得的使用而加速,这些证据较为支持第一种猜测。比较遗憾的是,患者在1981年首次出现症状,但直到大约25年之后才在临床普及对AQP4抗体的检测,因此在患者病程前期并未能完善该项检查。
另一种假设是,患者先后出现了两种不同的神经炎性疾病。尽管患者最初脊髓炎和脑干综合征均可出现在MS和NMOSD中,但使用皮质类固醇后这些症状几乎完全恢复,并且在之后的30年的时间中应用干扰素β-1a并未出现明显的疾病活动。随后,醋酸格拉替雷继续维持患者长达10年时间的病情相对稳定。从影像学上,颅内病变具有高度MS特征,包括脑室周围卵圆形高信号、Dawson手指征和脑萎缩。病程早期进行的脊髓成像显示多灶性和不连续的短节段病变,均符合明确的MS诊断标准。此外,85%的MS患者可见脑脊液寡克隆区带,而在NMOSD中寡克隆区带阳性仅占15%。
长时间的随访记录了患者从RRMS到SPMS的转变,同样符合MS特征,而并不符合NMOSD经典病程。然而,2019年中后期,患者开始在临床、影像学及实验室检测中显示出疾病活动方面的明显不同,包括距起病30多年后的长节段横贯性脊髓炎发作和AQP4抗体阳性,这种情况在MS中是高度不典型的。因此,似乎证据更支持第二种推断,也就是该患者先后出现了MS和NMOSD两种不同的神经系统炎性疾病。该病例提示神经内科医师在疾病出现不典型表现或者对药物反应欠佳时应及时重新审视疾病的诊断,以做出最合适的治疗方案。
参考文献
1.Goldschmidt C, Galetta SL, Lisak RP, Balcer LJ, Hellman A, Racke MK, Lovett-Racke AE, Cruz R, Parsons MS, Sattarnezhad N, Steinman L, Zamvil SS, Frohman EM, Frohman TC. Multiple Sclerosis Followed by Neuromyelitis Optica Spectrum Disorder: From the National Multiple Sclerosis Society Case Conference Proceedings. Neurol Neuroimmunol Neuroinflamm. 2022 Oct 21;10(1):e200037.
版权声明
如需转载或引用,须在醒目位置处注明“转自:罕见病新进展”。以上内容来自良医汇-罕见病新进展,如有疑问欢迎致电沟通:021-64360386。
 长按二维码
长按二维码关注精彩内容





